

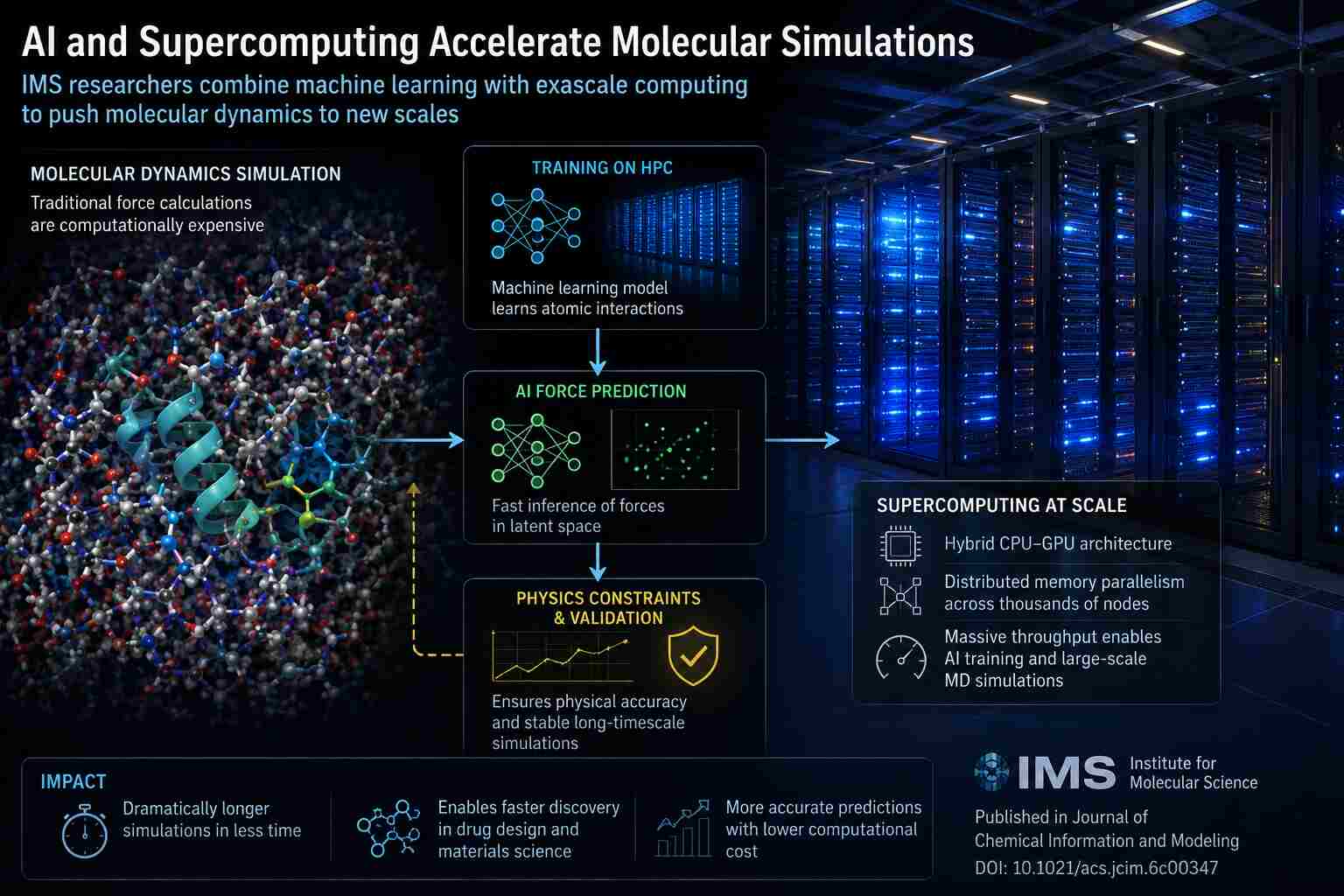
Scientists at Japan’s Institute for Molecular Science (IMS) have introduced a computational framework that dramatically speeds up atomistic molecular simulations by integrating machine learning with large-scale, high-performance computing systems. As detailed in the Journal of Chemical Information and Modeling, their research illustrates how AI-driven molecular dynamics is shifting from a specialized acceleration tool to a foundational element in the architecture of next-generation scientific computing.
The IMS team focused on one of computational chemistry’s longstanding bottlenecks: the enormous computational cost of accurately simulating molecular interactions across biologically relevant timescales. Conventional molecular dynamics (MD) simulations require repeated calculations of atomic forces over millions or billions of time steps, leading to exponential scaling as molecular systems grow in complexity. Even modern GPU clusters struggle to efficiently simulate large biomolecular systems with quantum-level accuracy.
The new framework addresses this limitation by integrating machine-learning-assisted force prediction into traditional MD pipelines. Instead of explicitly recalculating all interaction potentials at every timestep using computationally expensive physics-based methods, the AI system learns approximations of molecular force landscapes from prior simulation data. Once trained, the model can infer atomic interactions at dramatically lower computational cost while preserving high physical fidelity.
From a computer-science perspective, the architecture resembles a hybrid scientific inference engine operating across heterogeneous HPC infrastructure. Physics-based simulation kernels handle critical numerical stability constraints, while learned surrogate models accelerate portions of the computational workload that are traditionally dominated by expensive force-field evaluations.
This design reflects a broader transformation underway across supercomputing centers worldwide. Increasingly, exaflops scientific applications are adopting AI surrogates to reduce computational complexity in large-scale simulations. Rather than replacing numerical physics entirely, machine learning acts as an adaptive approximation layer capable of compressing otherwise intractable calculations.
The IMS researchers specifically targeted scalability challenges associated with high-dimensional molecular systems. Modern biomolecular simulations generate massive state spaces involving atomic coordinates, thermodynamic constraints, solvent interactions, and long-range electrostatic calculations. Traditional MD frameworks must continuously solve these interactions through iterative numerical integration methods, creating severe memory-bandwidth and floating-point throughput demands on HPC systems.
According to the published work, the researchers leveraged supercomputing resources to train and validate machine-learning models capable of accelerating molecular trajectory prediction without destabilizing the underlying simulation dynamics. This is computationally nontrivial because small force-prediction errors can compound over millions of timesteps, causing simulations to diverge from physically realistic behavior.
To address this, the framework integrates AI predictions with physics-informed constraints and validation stages. The result is a hybrid simulation architecture balancing computational acceleration against numerical stability, an increasingly important design pattern in scientific AI systems.
The project also highlights the growing importance of heterogeneous many-core architectures in computational chemistry. Modern MD workloads increasingly rely on tightly coupled CPU-GPU execution, distributed task scheduling, and multi-level parallelization strategies capable of scaling across thousands of compute nodes. Recent HPC studies in the Journal of Chemical Information and Modeling have demonstrated that optimized heterogeneous architectures can achieve near-linear scalability for large simulation workloads while dramatically reducing execution time for protein-ligand and biomolecular simulations.
For computer scientists, the significance extends beyond chemistry itself. Molecular simulation has become one of the most demanding benchmark domains for exaflops computing because it simultaneously stresses floating-point performance, memory locality, communication overhead, and distributed scheduling efficiency.
AI-enhanced MD frameworks such as the IMS system effectively transform simulation workloads into hybrid compute pipelines where numerical solvers and neural inference engines coexist inside the same execution stack. That convergence is reshaping both scientific software engineering and supercomputer architecture design.
The implications are especially important for pharmaceutical discovery and materials science. Traditional drug-discovery simulations often require months of compute time to evaluate protein folding, ligand binding, or molecular stability across sufficiently large sampling windows. AI-assisted acceleration could compress those timelines dramatically, enabling more iterative computational experimentation before laboratory validation begins.
The IMS work also aligns with a larger trend toward data-centric simulation environments. Molecular dynamics is no longer treated solely as a numerical integration problem. Increasingly, simulations generate streaming datasets suitable for real-time inference, optimization, and adaptive control systems. Emerging infrastructures are even beginning to treat simulation outputs as continuously queryable data services rather than static batch computations.
This evolution mirrors developments in climate modeling, astrophysics, and fusion-energy research, where AI-assisted surrogate modeling is becoming essential for managing computational complexity at exaflops scales.
For the supercomputing industry, the broader message is clear: the future of scientific computing may depend less on raw FLOPS growth and more on intelligent workload reduction through learned approximations. AI is increasingly being deployed not simply as an analytics layer operating after simulations complete, but as an active participant inside the simulation process itself.
This shift is redefining the purpose of supercomputers. Instead of serving solely as brute-force number-crunching machines, next-generation HPC platforms are evolving into adaptive computational ecosystems. In these environments, physics solvers, probabilistic inference engines, and machine-learning accelerators work seamlessly together as integrated elements within unified scientific workflows.